Basic Microbiome Analysis Tutorial
This is a basic microbiome analysis tutorial using AMPtk pipeline. This SOP/tutorial includes 1) processing raw sequence data files, 2) clustering/denoising sequences, and 3) taxonomy assignment. This tutorial dose not require installation, you can simply use Rstudio Cloud on your browser.
เว็บเพจนี้สอนวิธีการวิเคราะห์ข้อมูลความหลากหลายของจุลินทรีย์(ไมโครไบโอม)เบื้องต้น โดยผู้เรียนไม่ต้องดาวน์โหลดโปรแกรมลงบนคอมพิวเตอร์ส่วนตัว เพียงใช้ Rstudio Cloud การวิเคราะห์ข้อมูลไมโครไบโอมเบื้องต้นที่จะกล่าวถึงนั้น มี 3 ขั้นตอนหลัก คือ 1) processing raw sequence data files, 2) clustering/denoising sequences, และ 3) taxonomy assignment.
Step A: Open Rstudio cloud and Launch Terminal
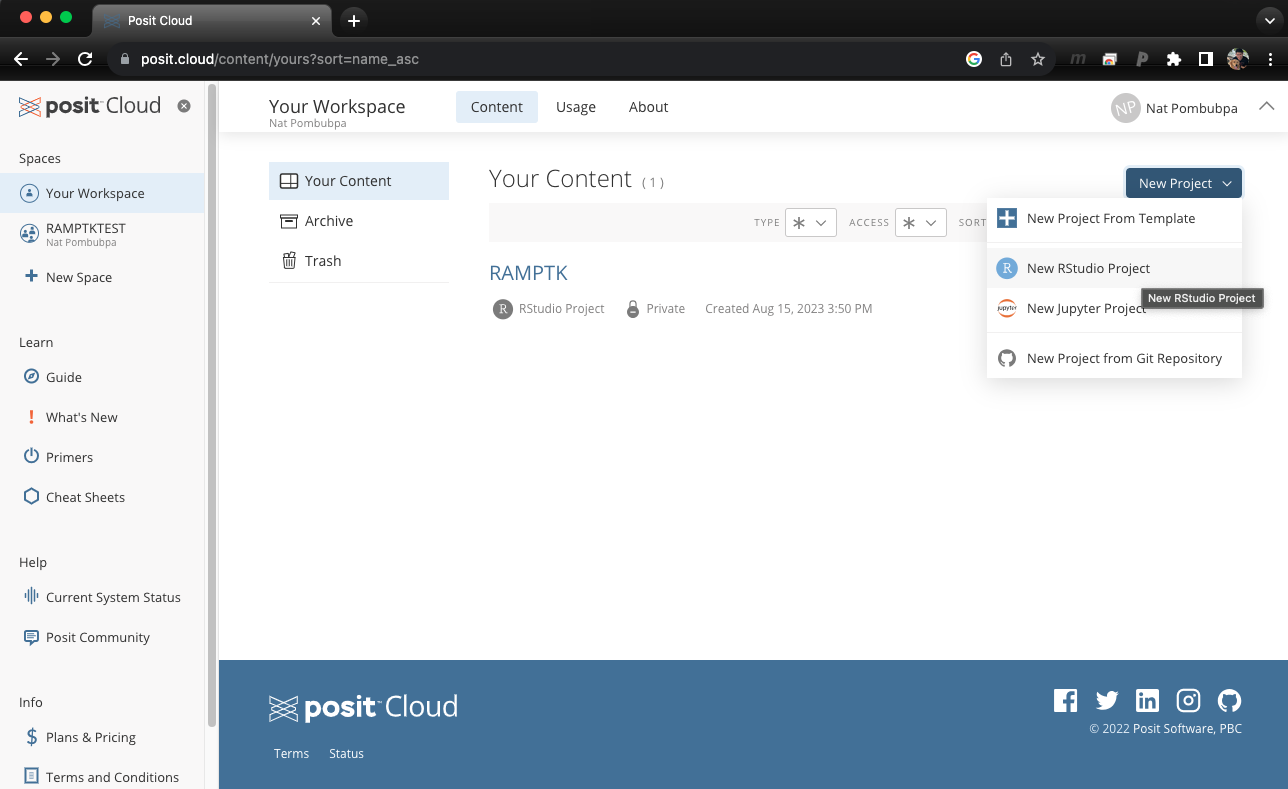
Once you log in to Rstudio cloud, your web browser should bring up a similar window as the picture shown above. Click the button on the top right corner to create a new Rstudio project. Then, the next step is to click “Terminal” which should look like a picture below after you click on it.
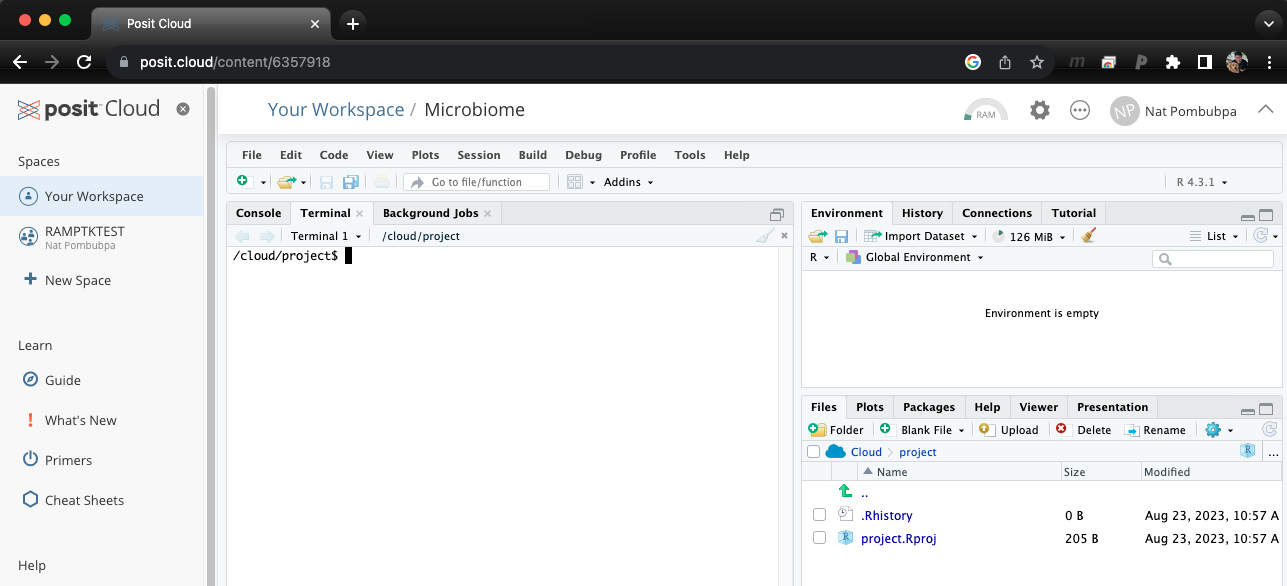
Step 1: Miniconda set up
# download and install miniconda3 to Rstudio cloud
/cloud/project$ wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
/cloud/project$ bash Miniconda3-latest-Linux-x86_64.sh
Check conda installation
/cloud/project$ conda env list
# If there's any issue, try this
/cloud/project$ source ~/.bashrc
Step 2: Install AMPtk
We will use AMPtk to process amplicon data.
#setup your conda env with bioconda, type the following in order to setup channels
(base) /cloud/project$ conda config --add channels defaults
(base) /cloud/project$ conda config --add channels bioconda
(base) /cloud/project$ conda config --add channels conda-forge
#create amptk env
(base) /cloud/project$ conda create -n amptk amptk
Next step is to verify that AMPtk can be used.
# simply type "amptk", amptk manual page should show up.
# amptk manual page is longer than this,
# but I only show you the first few lines.
(base) /cloud/project$ conda activate amptk
(amptk) /cloud/project$ amptk
Usage: amptk <command> <arguments>
version: 1.5.5
If everything work perfectly for you, you are almost ready for the actual analysis. Next step is to install database
(amptk) /cloud/project$ amptk install -i ITS
Check database installed
(amptk) /cloud/project$ amptk info
------------------------------
Running AMPtk v 1.5.5
------------------------------
Taxonomy Databases Installed: /home/r1200493/miniconda3/envs/amptk/lib/python3.10/site-packages/amptk/DB
------------------------------
DB_name DB_type FASTA Fwd Primer Rev Primer Records Source Version Date
ITS vsearch UNITE_public_all_10.05.2021.fasta ITS1-F ITS4 1389964 UNITE 8.3 2021-11-25
ITS1_SINTAX sintax sh_general_release_dynamic_s_all_10.05.2021_dev.fasta ITS1-F ITS2 140785 UNITE 8.3 2021-11-25
ITS2_SINTAX sintax sh_general_release_dynamic_s_all_10.05.2021_dev.fasta fITS7 ITS4 121896 UNITE 8.3 2021-11-25
ITS_SINTAX sintax sh_general_release_dynamic_s_all_10.05.2021_dev.fasta ITS1-F ITS4 161763 UNITE 8.3 2021-11-25
------------------------------
Step 3: Pre-processing/Demultiplexing
We can get data from a public dataset stored in NCBI. First let’s look at the BioProject page PRJNA659596. This page provides links to the 256 SRA experiments for 16S and ITS amplicon data from “Belowground Impacts of Alpine Woody Encroachment are determined by Plant Traits, Local Climate and Soil Conditions” project.
Although, this study has both 16S and ITS amplicon data, we will perform data processing only on several samples from ITS data.
 More detials on Sequencing setup can be found here
More detials on Sequencing setup can be found here
ITS primers for this project contain unique barcode for each sample. We usually submit ~200 samples per illumina miseq run. After sequencing process, the barcodes will be used to split sequences into fastq file for each sample.
Download tutorial data
(amptk) /cloud/project$ svn export https://github.com/NatPombubpa/Binder_Amptk_v1.4.2/trunk/illumina
Before we begin Pre-processing data, let’s take a look at our data.
#All data are in illunmina folder
#You should have 22 fastq files in this folder
(base) /cloud/project$ ls illumina/
ITS-cc-121_S26_L001_R1_001.fastq.gz ITS-cc-21_S123_L001_R2_001.fastq.gz
ITS-cc-121_S26_L001_R2_001.fastq.gz ITS-cc-221_S136_L001_R1_001.fastq.gz
ITS-cc-212_S126_L001_R1_001.fastq.gz ITS-cc-221_S136_L001_R2_001.fastq.gz
ITS-cc-212_S126_L001_R2_001.fastq.gz ITS-cc-27_S156_L001_R1_001.fastq.gz
ITS-cc-213_S127_L001_R1_001.fastq.gz ITS-cc-27_S156_L001_R2_001.fastq.gz
ITS-cc-213_S127_L001_R2_001.fastq.gz ITS-cc-76_S210_L001_R1_001.fastq.gz
ITS-cc-216_S130_L001_R1_001.fastq.gz ITS-cc-76_S210_L001_R2_001.fastq.gz
ITS-cc-216_S130_L001_R2_001.fastq.gz ITS-cc-91_S227_L001_R1_001.fastq.gz
ITS-cc-217_S131_L001_R1_001.fastq.gz ITS-cc-91_S227_L001_R2_001.fastq.gz
ITS-cc-217_S131_L001_R2_001.fastq.gz ITS-cc-98_S234_L001_R1_001.fastq.gz
ITS-cc-21_S123_L001_R1_001.fastq.gz ITS-cc-98_S234_L001_R2_001.fastq.gz
What does the sequence file look like?
(amptk) /cloud/project$ zmore illumina/ITS-cc-121_S26_L001_R1_001.fastq.gz | head -2
@M02457:311:000000000-C6VB2:1:1101:18927:1862 1:N:0:ATGTCCAG+CAGTCGGA
AGCCTCCGCTTATTGATATGCTTAAGTTCAGCGGGTGGTCCTACCTGATTTGAGGTCAGAGTCCAAAAGAGCGCCACAAGGGGCAGGTTATGAGCGGGCCTCACACCATGCCAGACGAAACTTATCACGTCAGGACGTGGATGCTGGTCCCACTAAGTCATTTGAGGCAAGCCGGCAGACGGCAGACACCCAGGTCCATGTCCACCCCAGGTCAAGGAGACCCGAGGGGATTGAGATTTCATGACACTCAAACAGGCATGCCTTTCGGAATACCAAAAGGCGCAAGGTGCGTTCGAAGATT
Now, we can begin Pre-processing steps, BUT……!!!
There are several different file format that could be generated from Illumina Miseq sequencing (or sequencing centers). We’ll focus on demultiplexed PE reads in which all the sequences were splited into separated fastq files for each samples. The general workflow for Illumina demultiplexed PE reads is:
- Merge PE reads (use USEARCH or VSEARCH)
- filter reads that are phiX (USEARCH)
- find forward and reverse primers (pay attention to –require_primer argument)
- remove (trim) primer sequences
- if sequence is longer than –trim_len, truncate sequence

#Pre-preocessing steps will use `amptk illumia` command for demultiplexed PE reads
(amptk) /cloud/project$ amptk illumina -i illumina/ -f AACTTTYRRCAAYGGATCWCT -r AGCCTCCGCTTATTGATATGCTTAART --require_primer off -o MicroEco --rescue_forward on --primer_mismatch 2 -l 250
Step 4: Clustering

This step will cluster sequences into Operational Taxonomy Unit (OTU), then generate representative OTU sequences and OTU table. OTU generation pipelines in AMPtk uses UPARSE clustering with 97% similarity (this can be changed).
Note: at clustering step, we used merged sequence from STEP1 as an input and we will generate clustered sequences file and OTU table.
(amptk) /cloud/project$ amptk cluster -i MicroEco.demux.fq.gz -o MicroEco
Step 5: Taxonomy assignment
This step will assign taxonomy to each OTU sequence and add taxonomy to OTU table. This command will generate taxnomy based on the ITS database.
Note: at Taxonomy Assignment step, we will use clustered sequences file and OTU table for taxonomy assignment from ITS database
(base) /cloud/project$ amptk taxonomy -f MicroEco.cluster.otus.fa -i MicroEco.otu_table.txt -d ITS --method sintax
When the taxonomy assignment is completed, we can check the taxonmy file.
# select Fungi from taxonomy file by using grep command
(amptk) /cloud/project$ grep Basidiomycota MicroEco.cluster.taxonomy.txt | head -10
OTU34 SINTAX;k:Fungi,p:Basidiomycota,c:Microbotryomycetes,f:Chrysozymaceae,g:Slooffia
OTU37 SINTAX;k:Fungi,p:Basidiomycota,c:Microbotryomycetes,f:Chrysozymaceae,g:Slooffia
OTU236 SINTAX;k:Fungi,p:Basidiomycota,c:Agaricomycetes,o:Agaricales,f:Inocybaceae,g:Inocybe
OTU307 SINTAX;k:Fungi,p:Basidiomycota,c:Agaricomycetes,o:Thelephorales,f:Thelephoraceae
OTU394 SINTAX;k:Fungi,p:Basidiomycota,c:Agaricomycetes,o:Agaricales,f:Inocybaceae,g:Inocybe
OTU470 SINTAX;k:Fungi,p:Basidiomycota,c:Agaricomycetes,o:Thelephorales,f:Thelephoraceae
OTU587 SINTAX;k:Fungi,p:Basidiomycota,c:Agaricomycetes,o:Thelephorales,f:Thelephoraceae
OTU648 SINTAX;k:Fungi,p:Basidiomycota,c:Agaricomycetes,o:Cantharellales,f:Clavulinaceae,g:Clavulina
OTU665 SINTAX;k:Fungi,p:Basidiomycota,c:Microbotryomycetes,f:Chrysozymaceae,g:Slooffia
OTU688 SINTAX;k:Fungi,p:Basidiomycota,c:Agaricomycetes,o:Agaricales,f:Cortinariaceae,g:Cortinarius