Basic Unix and Version control for Biologists EP3 is aiming to helps anyone who would like to learn basic unix programming and version control using github. This introduction/tutorial dose not require installation, you can simply click you can simply use Rstudio Cloud on your browser.
เว็บเพจนี้สอน Unix Shell เบื้องต้น โดยผู้เรียนไม่ต้องดาวน์โหลดโปรแกรมลงบนคอมพิวเตอร์ส่วนตัว เพียงใช้ Rstudio Cloud บนเว็บบราวเชอร์
Open Binder and Launch Terminal
Step A: Open Rstudio cloud and Launch Terminal
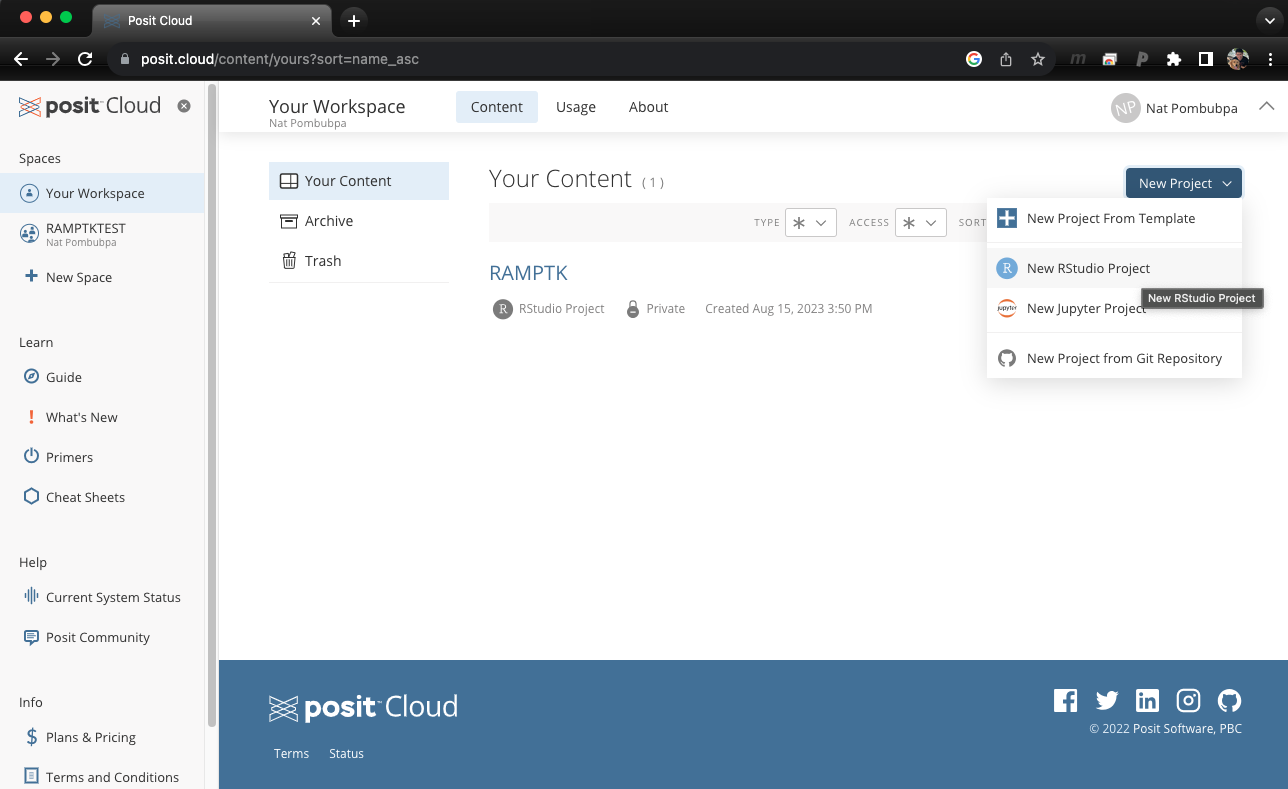
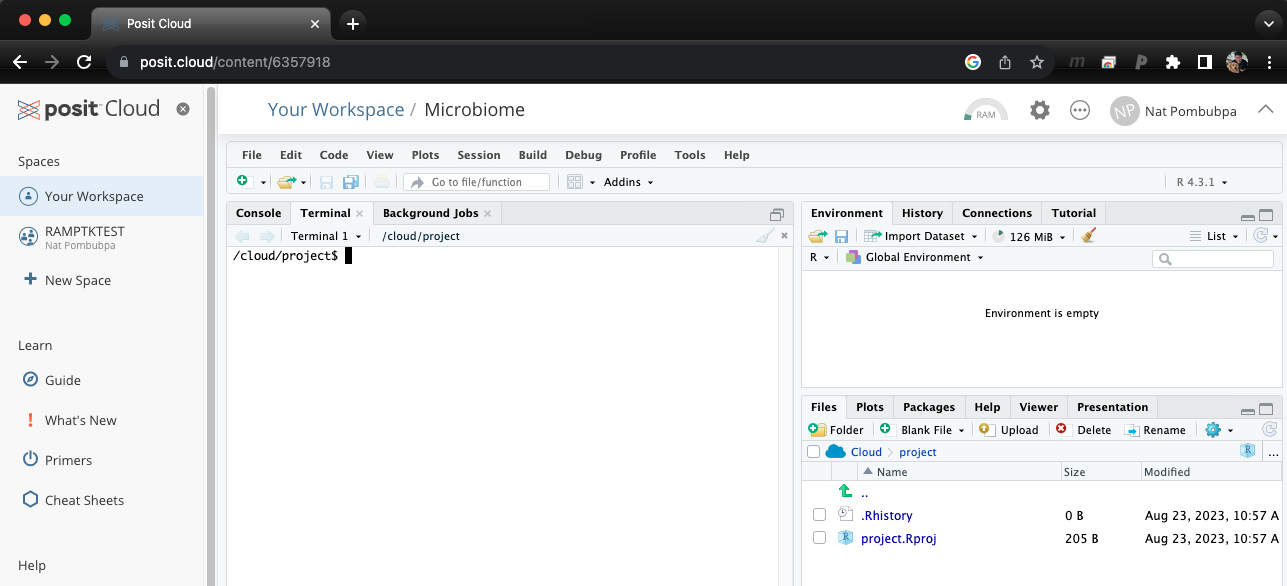
Once you log in to Rstudio cloud, your web browser should bring up a similar window as the picture shown above. Click the button on the top right corner to create a new Rstudio project. Then, the next step is to click “Terminal” which should look like a picture below after you click on it.
Hands-On Exercise (45 minutes)
- Downloading a Sample Dataset
- Example: FASTA file of DNA sequences
- Processing the Data
- Viewing the data structure
- Counting sequences
- Extracting specific sequences using
grep
Creating Bash Scripts and Version Control (60 minutes)
- Creating Bash Scripts
- Writing and executing a simple script
- Version Control with GitHub
- Basic Git commands
- Pushing code to GitHub
Summary and Q&A (45 minutes)
- Recap of Key Commands
- Real-World Applications in Bioinformatics
- Questions and Discussion
Lesson
Hands-On Exercise (45 minutes)
Basic command line recap Downloading a Sample Dataset
- Download a sample FASTA file of DNA sequences.
/cloud/project$ wget -O SRR21388484.fasta.gz https://trace.ncbi.nlm.nih.gov/Traces/sra-reads-be/fasta?acc=SRR21388484
Processing the Data
- View the structure of the FASTA file using
cat,less, andhead.
/cloud/project$ gunzip SRR21388484.fasta.gz
/cloud/project$ cat SRR21388484.fasta | head
/cloud/project$ less SRR21388484.fasta
note: type q to quit less
/cloud/project$ head SRR21388484.fasta
- Count the number of sequences in the file using
grepandwc.
/cloud/project$ grep -c ">" SRR21388484.fasta
/cloud/project$ grep ">" SRR21388484.fasta | wc -l
Creating Bash Scripts and Version Control (60 minutes)
Creating Bash Scripts
A bash script is a file containing a series of commands that you can execute together. Here’s how to create and execute a simple bash script:
- Create a new file called
script.sh:
/cloud/project$ vim script.sh
- Add the following lines to the script:
#!/bin/bash
# This script prints the current date and lists files in the directory
echo "Current date and time:"
date
echo "Files in the current directory:"
ls
save and exit by type :wq and hit Enter
- Execute the script:
/cloud/project$ bash script.sh
Version Control with GitHub
Version control is essential for tracking changes to your code and collaborating with others. Here’s how to use Git and GitHub for version control:
- Create an account on Github
- Create a New Repository on GitHub:
- Go to GitHub and create a new repository.
- we’ll demonstrate this on github.
Additional Topics
Working with FASTQ Files
FASTQ is a common file format used to store sequences and their corresponding quality scores in bioinformatics. Understanding how to work with FASTQ files is essential for tasks like analyzing high-throughput sequencing data.
Introduction to the FASTQ Format:
FASTQ files typically contain information about DNA or RNA sequences obtained from next-generation sequencing (NGS) platforms. Each record in a FASTQ file consists of four lines:
- Sequence Identifier (ID): Begins with “@” and contains information about the read or sequence.
- Sequence Data: Contains the actual DNA or RNA sequence as a string of characters (A, C, G, T, or N).
- Quality Identifier (ID): Begins with “+” and is often the same as the sequence identifier.
- Quality Scores: Represents the quality of each base in the sequence as ASCII characters.
Here’s an example FASTQ record:
/cloud/project$ wget -O SRR21388484.fastq.gz https://trace.ncbi.nlm.nih.gov/Traces/sra-reads-be/fastq?acc=SRR21388484
/cloud/project$ gunzip SRR21388484.fastq.gz
/cloud/project$ head SRR21388484.fastq
@SRR21388484.1 1 length=301
TACGTAGGGGGCGAGCGTTGTCCGGAATTATTGGGCGTAAAGAGCGTGTAGGCGGTTTGGTAAGTCTGCC
GTGAAAACCCGGGGCTCAACCCCGGTCGTGCGGTGGATACTGCCAGGCTAGAGGATGGTAGAGGCGAGTG
GAATTCCCGGTGTAGCGGTGAAATGCGCAGATATCGGGAGGAACACCAGTAGCGAAGGCGGCTCGCTGGG
CCATTCCTGACGCTGAGACGCGAAAGCTAGGGGAGCGAACAGGATTAGATACCCTGGTAGTCCGGCTGAC
TGACTATCTCGTATTCCGTCT
+SRR21388484.1 1 length=301
??????????????????????????????????????????????????????????????????????
??????????????????????????????????????????????????????????????????????
??????????????????????????????????????????????????????????????????????
Quality score refrence https://help.basespace.illumina.com/files-used-by-basespace/quality-scores
Viewing FASTQ Files:
- Use
catto display the entire file on the terminal (suitable for small files).
/cloud/project$ cat SRR21388484.fastq | head -5
- Use
lessfor interactive viewing of large FASTQ files.
/cloud/project$ less SRR21388484.fastq
- Use
morefor interactive viewing similar toless.
/cloud/project$ more SRR21388484.fastq
Combining, Splitting, and Converting File Formats:
- Split a large FASTQ file into smaller files based on the number of records:
/cloud/project$ split -l 600006 SRR21388484.fastq 600kSRR
- Combine multiple FASTQ files into one:
/cloud/project$ cat 600kSRRaa 600kSRRab > combined.fastq
Extracting Sequences from FASTQ Files Based on IDs:
- Use
grepto extract sequences based on their sequence identifiers:
/cloud/project$ vim sequence_ids.txt
sequence_ids.txtshould contain one sequence ID per line:, try addingSRR21388484.1andSRR21388484.2
/cloud/project$ grep -A 3 -f sequence_ids.txt combined.fastq > extracted_sequences.fastq
Example
@seq_id_1
@seq_id_2
...
/cloud/project$ head extracted_sequences.fastq
@SRR21388484.1 1 length=301
TACGTAGGGGGCGAGCGTTGTCCGGAATTATTGGGCGTAAAGAGCGTGTAGGCGGTTTGGTAAGTCTGCC
GTGAAAACCCGGGGCTCAACCCCGGTCGTGCGGTGGATACTGCCAGGCTAGAGGATGGTAGAGGCGAGTG
GAATTCCCGGTGTAGCGGTGAAATGCGCAGATATCGGGAGGAACACCAGTAGCGAAGGCGGCTCGCTGGG
--
+SRR21388484.1 1 length=301
??????????????????????????????????????????????????????????????????????
??????????????????????????????????????????????????????????????????????
??????????????????????????????????????????????????????????????????????
--
Summary and Q&A (45 minutes)
- Recap the key UNIX commands covered.
- Discuss how these commands are applied in real-world bioinformatics.
- Open the floor for questions and further discussion.